Medical Device Identification
FDA Mandate: Unique Device Identifiers (UDI)
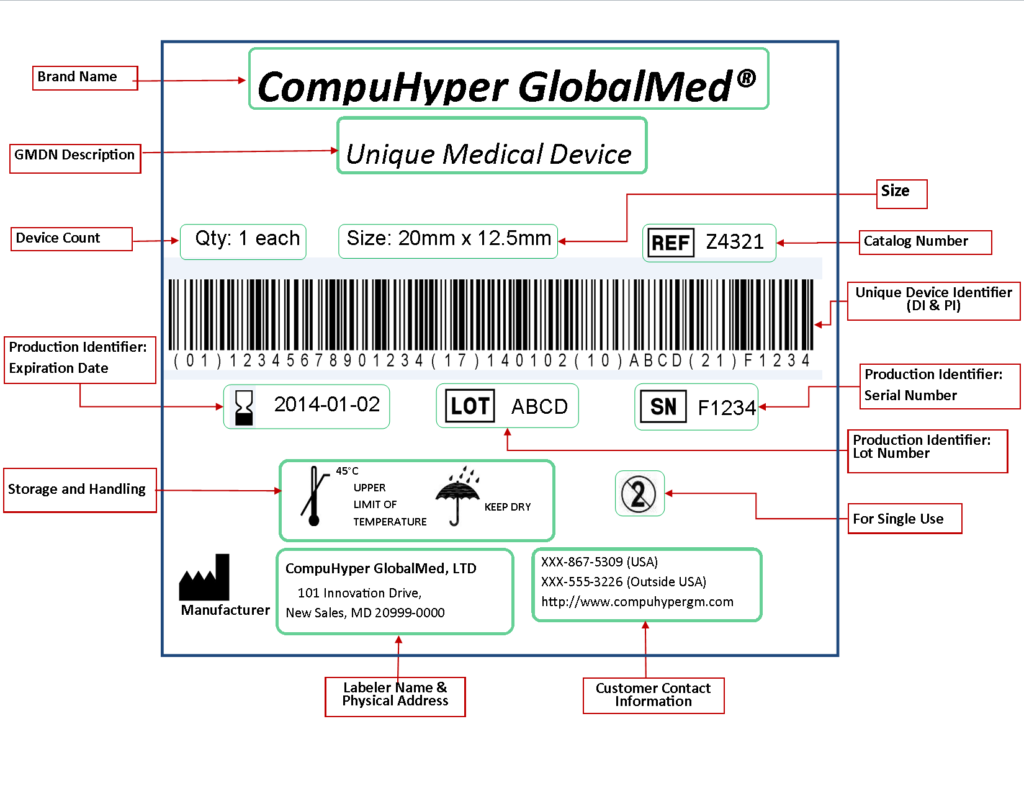
The Food and Drug Administration (FDA) of the United States has devised a unique device identification system to properly identify medical devices during their distribution and usage. Most devices' labels will include a unique device identifier (UDI) in both human and machine-readable formats when fully implemented. Device labelers must additionally provide specific device information to the FDA's Global Unique Device Identification Database (GUDID). The public can use AccessGUDID to look for and download FDA information.
The UDI system, which was implemented in 2013, provides a variety of benefits that will be realized more completely with the acceptance and integration of UDIs into the healthcare delivery system. According to the FDA, UDI deployment will improve patient safety, modernize device post-market surveillance, and make medical device innovation easier.
Who Needs To Comply?
The FDA UDI rule applies to a device labeler. A labeler is anyone who applies a label to a device or modifies a device's label with the aim that the device would be commercially disseminated without any later replacement or modification of the label. The addition of the name and contact information of a person who distributes the device without making any other changes to the label is not considered a modification for establishing whether a person is a labeler.
The labeler is typically the device maker, but it can also be a specification developer, a single-use device reprocessor, a convenience kit assembler, a repackager, or a relabeler.
What is a Unique Device Identifier?
A unique device identifier (UDI) is a unique numeric or alphanumeric code that generally consists of the following:
- Device Identifier (DI), a mandatory, fixed portion of a UDI that identifies the labeler and the specific version or model of a device.
-
Production identifier (PI), a conditional, variable portion of a UDI that identifies one or more
of the following when included on the label of a device:
- Lot or batch number within which a device was manufactured
- Serial number of a specific device
- The expiration date of a specific device
- Date a specific device was manufactured
- Distinct identification code required by §1271.290(c) for a human cell, tissue, or cellular and tissue-based product (HCT/P) regulated as a device
The device labeler must provide the UDI in two forms on labels and packages:
- Easily readable plain-text
- Machine-readable form that uses automatic identification and data capture (AIDC) technology
FDA-Accredited Issuing Agencies
The keyword in the phrase "Unique Device Identifier" is unique. Rather than selecting a single industry numbering agency to manage the label identifiers and adhere to a single numbering model, the FDA certified 3 existing agencies. All three already had sects of companies abiding by their standards and identifiers, so this provided a comprehensive solution for companies.
The accredited issuing agencies are: GS1, HIBBC (Health Industry Business Communication Council), and ICCBBA.
Sample Label

Courtesy of www.usa.philips.com